13年前
一个小生命在福建泉州安溪呱呱坠地
但黄爸爸(化名)的疑问却随之而来
“我家娃娃,究竟是儿子还是女儿?”
说是儿子吧
可染色体检查结果却是“XX”
说是女儿吧
看起来却是个“带把儿的”娃娃
整得当地的医生们也一头雾水
但由于家庭贫困,黄爸爸无法带孩子进行进一步的检查和治疗,令他没想到的是,几年后,当第二个孩子出生时,同样的怪事又再次发生了!
这些年,黄爸爸辗转多家医院,但俩孩子的问题还是没取得实质性解决。眼看着大娃小婷(化名)进入青春期后“男子气概”一天天明显,二娃小芬(化名)也已经到了上小学的年级,一家人抱着一丝的希望,来到了福建医科大学附属第一医院就诊。
男女难辨 众人携手查病因
去年暑假,福建医科大学附属第一医院小儿外科许雅丽副主任医师接诊了这一对特殊的姐妹。经与性发育异常多学科会诊(DSD MDT)团队讨论,并对其展开了一系列针对性检查,发现两姐妹从遗传学角度而言,确实是女性,有卵巢、子宫等器官。
但体格检查两人的男性化情况较为严重,其中小婷正值青春,皮肤黝黑,身材矮小,有喉结,声音浑厚,看外表是一个典型的“小子”。
此外,外生殖器检查中发现其有严重的阴蒂肥大,大小和成年男性差不多,伴有阴道尿道融合,外观看只有一个开口,(Prader分级2-3级)。
综合种种情况,医生们脑海中逐渐浮现出了一个词:异常高雄激素分泌。
又经过一系列检查,排除了各种诱因后,医生们判断她们患的是“先天性肾上腺皮质增生症”这个常染色体隐性遗传病。
药物手术双管齐下
姐妹俩恢复“女儿身”
病因找到了,那要怎么治疗呢?
附一小儿外科DSD MDT团队综合评估姐妹俩临床表型、遗传内分泌和性心理等整体情况,制定了个体化的治疗方案,先进行术前的内分泌治疗,把激素逐渐降低至正常水平后,再通过手术重建生殖器。
小儿外科主任林俊山主任医师随即带领团队评估得出两人手术的方向是恢复“女儿身”。
由于姐妹俩阴道发育不良,尿道和阴道融合,共同通道长度仅为3厘米,给手术带来了一定的挑战。
好在手术很顺利。待到出院时,外生殖器已和正常女性一般无二,且无明显瘢痕。
这种罕见病其实可以预防
据悉,先天性肾上腺皮质增生症(CAH)是一种罕见的由于基因突变引起肾上腺类固醇合成途径中的酶缺失,导致肾上腺皮质增生的常染色体隐性遗传代谢病。在新生儿中,发病率为5-10/10万。
有些患病的宝宝初期和健康宝宝没有太大区别,但后期会出现一系列身材矮小、男性化等表现。最危险的“失盐型”宝宝容易诱发高钾血症、低钠血症,很多宝宝在没有确诊时,由于得不到正确治疗,就遗憾夭折。
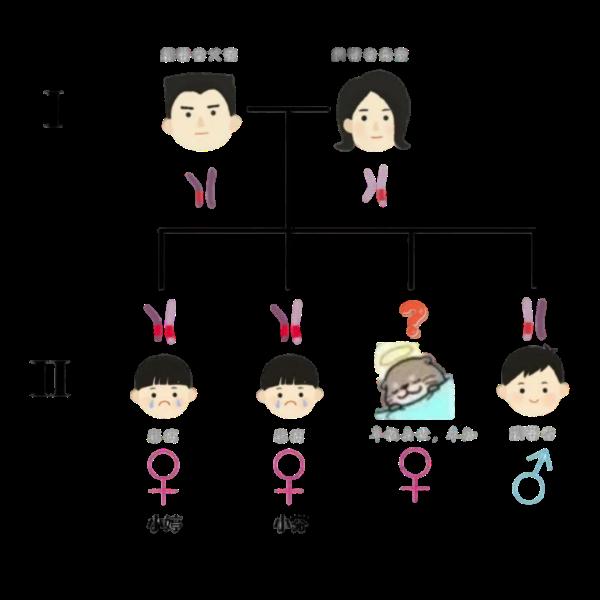
CAH是可防可治的一类疾病,需要“早发现,早诊断,早治疗”。在小婷一家的基因检测中发现,小婷的弟弟就仅是该致病基因的携带者。
如何预防?
因此,若夫妻双方有一方确定为致病基因的携带者,另一方就应该在孕前筛查是否携带与患者相同的致病基因,并到有相关资质的医疗机构接受遗传咨询。
若已怀孕,可在产前对胎儿进行基因检测,检测结果可作为是否继续妊娠的重要参考,也可应用试管婴儿体外基因诊断技术选择正常的胚胎后再植入母体子宫继续妊娠。